


Understanding Lipid Metabolism in Chickens and Where It Can Go Wrong

Dr. Nilay Deshpande1, Dr. Saurabh Mane2
1PhD Poultry Science, ICAR-Directorate of Poultry Research, Hyderabad
1MVSc Poultry Science, ICAR-Indian Veterinary Research Institute, Izzatnagar
Lipids represent one of poultry biology’s greatest paradoxes — simultaneously essential for optimal productivity and catastrophically dangerous when metabolism dysregulates. Modern chicken strains, refined through decades of genetic selection for explosive growth and extraordinary productivity, possess lipid metabolic machinery operating at remarkable efficiency. Yet this very efficiency, coupled with the metabolic stress of high-density production, creates a precarious system vulnerable to dysregulation. Understanding how chickens process dietary lipids—and critically, what happens when this process fails—is fundamental to contemporary poultry science. Lipids contribute over twice the energy per gram compared to proteins or carbohydrates, provide essential polyunsaturated fatty acids (omega-3 and omega-6) vital for immune competence and reproduction, and serve as carriers for fat-soluble vitamins A, D, E, and K. Yet when lipid metabolism spirals out of control, the consequences are severe: fatty liver syndrome, fatty liver haemorrhagic syndrome (FLHS), fatty liver kidney syndrome (FLKS), and hepatitis collectively represent one of the most significant challenges in modern poultry production.
From Ingestion to Hepatic Processing: The Initial Lipid Journey
The lipid metabolic odyssey begins in the gastrointestinal tract. Dietary triglycerides, the predominant lipid form in poultry feeds, undergo enzymatic hydrolysis by pancreatic lipase in the small intestine, yielding monoglycerides and free fatty acids. Bile acids emulsify these hydrophobic molecules, facilitating their incorporation into micelles that traverse the intestinal epithelium with impressive efficiency—typically 85-90% digestibility. Once absorbed, enterocytes re-esterify these components into triglycerides and package them into protomicrons— lipoprotein particles analogous to mammalian chylomicrons. These protomicrons enter the portal circulation, delivering absorbed lipids directly to the hepatocyte, establishing the liver as the metabolic epicentre determining the fate of dietary lipids: oxidation for energy, incorporation into structural membranes, or re-export to peripheral tissues. The composition of dietary lipid sources profoundly influences downstream metabolic consequences. Plant oils (soybean, sunflower, canola) provide predominantly linoleic acid (omega-6 PUFA) and oleic acid (MUFA), while animal fats contribute greater quantities of saturated and monounsaturated fatty acids.
The omega-3 to omega-6 ratio fundamentally shapes the lipid mediator profile—excessive omega-6 without compensatory omega-3 supplementation shifts the lipid-derived inflammatory mediator balance toward pro-inflammatory species, predisposing to metabolic dysfunction. Critically, chickens cannot synthesize linolenic acid, creating an absolute dietary requirement for this omega-3 PUFA.
Hepatic Synthesis and Export: The Metabolic Bottleneck
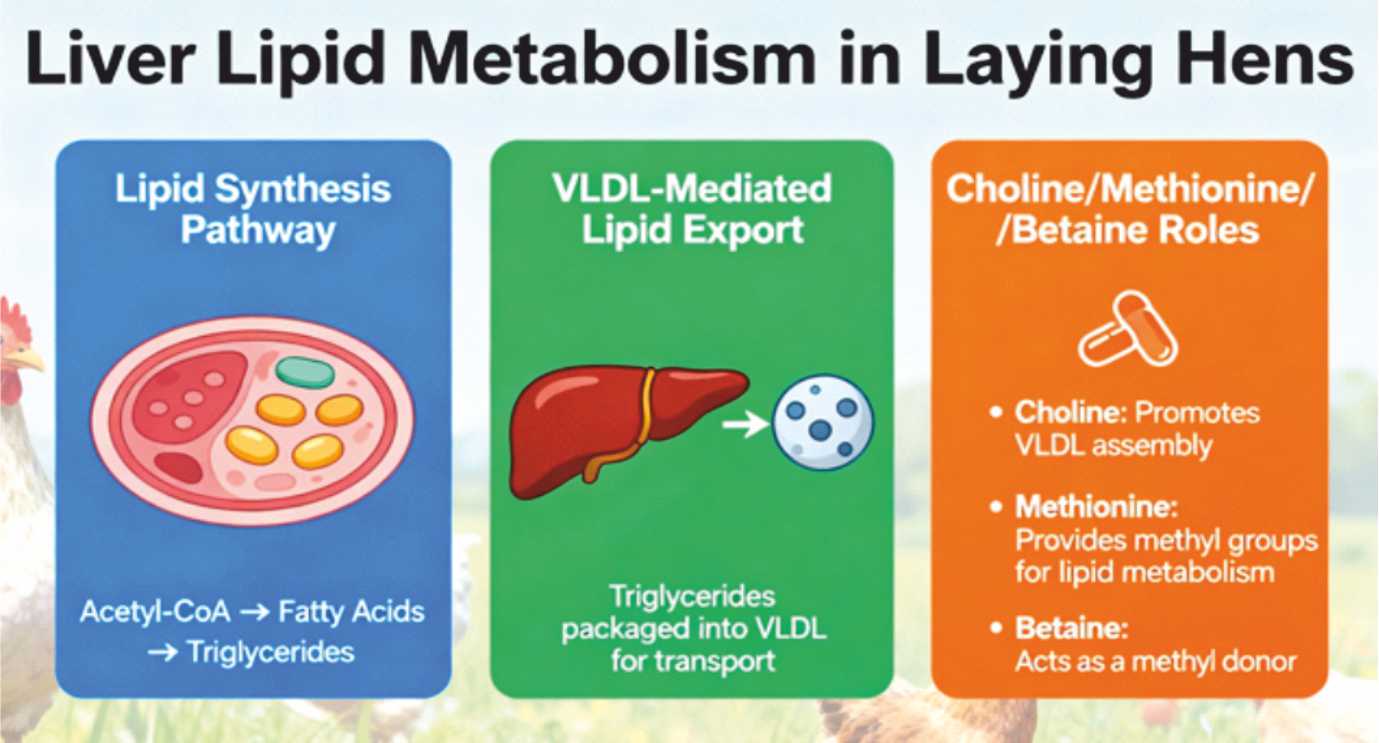
The liver functions simultaneously as processor of absorbed lipids and de novo fatty acid synthetic factory. In laying hens, the hepatic lipogenic capacity is extraordinary— synthesizing sufficient triglycerides to support daily yolk deposition, where lipids constitute approximately 33% of yolk mass by weight. This synthetic machinery operates through acetyl-CoA carboxylase and fatty acid synthase, generating novel fatty acids from carbon skeletons derived from dietary carbohydrates or amino acids. These newly synthesized lipids, together with absorbed dietary fatty acids, must be exported from hepatocytes to peripheral tissues —predominantly through very low-density lipoprotein (VLDL) particles and, in laying hens, through vitellogenin-mediated transport to the ovary.

The efficiency of hepatic lipid export fundamentally depends upon apolipoprotein synthesis, particularly apolipoprotein B (apoB), which serves as the structural scaffold of VLDL particles. This apoB synthesis, in turn, requires abundant phospholipid availability, which depends on choline—a nutrient that must be provided dietarily or synthesized through dietary methionine via methylation reactions. The lipotropic hypothesis elegantly explains why supplemental choline, methionine, and betaine mitigate fatty liver development: these nutrients are not direct energy sources but rather essential cofactors enabling the synthetic machinery supporting VLDL assembly and hepatic lipid export. When lipotropic substances become limiting, the hepatocyte becomes an anatomical traffic jam: lipids accumulate internally faster than export machinery can mobilize them peripherally, creating the pathological lipid accumulation characteristic of fatty liver.
When Export Fails: Pathophysiology of Fatty Liver and FLHS
Hepatic steatosis—excessive hepatic triglyceride accumulation—arises when hepatocyte lipid uptake and synthesis exceed oxidation and export capacity. In laying hens, this dysregulation commonly emerges from the synergistic dysfunction of multiple regulatory pathways. High-energy or high-fat diets, particularly those rich in saturated animal fats, overwhelm export capacity through sheer substrate excess. Simultaneously, inadequate lipotropic nutrient provision cripples VLDL assembly. The gene regulatory landscape becomes progressively dysregulated: the peroxisome proliferator-activated receptors (PPARα and PPARγ), which normally enhance fatty acid oxidation and promote metabolic flexibility, show reduced hepatic expression, while sterol regulatory element-binding protein 1 (SREBP1), a master transcription factor governing lipogenic enzyme expression, becomes hyperactivated. The consequence is a metabolic phenotype characterized by relentless lipogenesis coupled with suppressed lipolysis.
Fatty liver hemorrhagic syndrome represents the catastrophic progression of unchecked hepatic steatosis. Beyond simple triglyceride accumulation, FLHS involves severe impairment of VLDL secretion accompanied by oxidative stress, hepatocellular ballooning, and inflammatory cell infiltration. The accumulated lipids generate reactive oxygen species (ROS) as mitochondria become overwhelmed processing fatty acid substrates through β-oxidation. The hepatocellular accumulation of lipid droplets physically displaces functional hepatocytes, reducing synthetic capacity for essential proteins (albumin, clotting factors, cytochromes P450) and impairing detoxification function. Bile acid synthesis and signaling become dysregulated, further compromising lipid export. Ultimately, hepatic capillary rupture causes hemorrhage, often precipitating sudden mortality during capture or handling.
FLHS epidemiology reveals particularly severe disease manifestations in caged laying hens during peak productivity—the combination of extreme hepatic lipogenic demand, minimal physical activity reducing fatty acid oxidation, and often suboptimal nutritional management creates a metabolic catastrophe. Prevention requires aggressive intervention: dietary fat restriction to 3-5%, polyunsaturated fat emphasis (soybean oil 2-3%), omega-3 supplementation (flaxseed or fish oil 0.5-1%), and robust lipotropic provision (choline 1200-1500 ppm, methionine and betaine at NRC-recommended levels). Antioxidant fortification with vitamin E (100+ IU/kg) and selenium (0.3-0.5 ppm) protects hepatocytes from oxidative damage.
FLKS: The Young Broiler’s Metabolic Crisis
Fatty liver kidney syndrome predominantly affects rapidly-growing broiler chicks (2-6 weeks age), representing a distinct but equally severe lipid metabolism dysregulation. FLKS manifests as simultaneous pathological lipid accumulation in both liver and kidneys, precipitating growth depression, poor feed efficiency, and substantial mortality.
The pathophysiological substrate differs from FLHS: young broilers experience extraordinary anabolic demand for lipids required for cell membrane synthesis and organ development during rapid tissue accretion. The hepatic export system, dependent on lipotropic nutrient availability and coordinated gene expression, becomes rate-limiting under this intense metabolic stress.
Choline emerges as the critical intervention point. Deficiency impairs both phospholipid synthesis (necessary for VLDL assembly) and apoB expression, directly constraining VLDL particle formation. The resulting lipid entrapment in hepatocytes, combined with dysregulated lipid transport to peripheral tissues, precipitates renal lipid accumulation through mechanisms not yet fully elucidated—potentially involving impaired renal lipid oxidation capacity or inflammatory responses to elevated circulating lipid levels. Management requires elevated choline provision (800-1200 ppm), particularly in starter diets, combined with polyunsaturated fat inclusion (soybean, sunflower oils 3-5%) and comprehensive antioxidant protection. Notably, excessive dietary energy density paradoxically increases FLKS risk—high carbohydrate-based energy triggers amplified de novo hepatic lipogenesis, overwhelming export capacity.
Hepatitis and Metabolic Dysfunction: Inflammation Disrupts Lipid Homeostasis
Hepatitis—whether triggered virally, bacterially, toxically, or metabolically—fundamentally disrupts lipid homeostasis through multiple mechanisms. Hepatocellular inflammation directly impairs VLDL synthesis capacity, causing triglyceride and non-esterified fatty acid accumulation. Oxidative stress accompanying inflammation damages lipid membranes through peroxidation, generating lipid peroxides that perpetuate cellular damage. Heat stress-associated hepatitis particularly dysregulates lipid-related gene expression, specifically reducing PPARα and fatty acid oxidation capacity while maintaining or elevating lipogenic gene expression. The net result is secondary steatosis superimposed upon acute inflammation.
Dietary management during hepatitis requires omega-3 enrichment (fish oil 1-2%) to support synthesis of pro-resolving lipid mediators (lipoxins, resolvins, protectins) that actively terminate inflammation. Saturated fat restriction minimizes pro-inflammatory lipid mediator generation. Comprehensive antioxidant support—emphasizing natural vitamin E (80-100 IU/kg), selenium (0.3+ ppm), and potentially additional antioxidants—counters oxidative stress. Identification and elimination of the hepatitis trigger (viral vaccination, bacterial antimicrobials or probiotics, mycotoxin removal) remains paramount.
Integrated Prevention: Synthesizing Metabolic Knowledge into Practice
Effective prevention of lipid metabolism disorders requires systematic integration of nutritional, environmental, and managerial strategies. Dietary fat inclusion must balance energy requirements against metabolic risk—typically 3-5% for layers, 4-6% for broilers—with stringent prioritization of polyunsaturated plant oils over saturated animal fats. Lipotropic nutrient provision should exceed minimum requirements during stress periods: choline 1200-1500 ppm, methionine and betaine at NRC recommendations or above. Micronutrient fortification with vitamin E (50-100 IU/kg) and selenium (0.3+ ppm) protects against oxidative stress inherent to lipid-intensive metabolism.
Environmental management—heat stress mitigation through ventilation, optimal stocking densities permitting normal activity, biosecurity preventing stress-inducing pathogens—directly supports metabolic resilience. Feed quality monitoring ensuring absence of rancid fats and mycotoxins prevents additional hepatic burden. Strain-specific considerations recognize that modern high-productivity genetics carry metabolic vulnerabilities requiring targeted nutritional support; consultation with poultry nutritionists familiar with your specific genetic line optimizes intervention strategies.
Conclusion: Metabolic Excellence Through Informed Management
Lipid metabolism in chickens exemplifies the exquisite complexity underlying productive physiology—a system of extraordinary sophistication vulnerable to dysregulation under contemporary production stresses. The disorders arising from lipid metabolic failure—fatty liver, FLHS, FLKS, hepatitis—represent not arbitrary diseases but rather predictable consequences of pushing metabolism to or beyond biological limits. These conditions remain largely preventable through evidence-based nutritional and management practices informed by mechanistic understanding of underlying pathophysiology. By maintaining optimal dietary lipid balance emphasizing unsaturated sources, providing robust lipotropic and micronutrient support, managing environmental stressors, and employing strain-appropriate protocols, producers can sustain the metabolic machinery enabling both productivity and welfare. The lipid paradox—that lipids are simultaneously essential and potentially catastrophic—demands perpetual vigilance and informed decision-making; the reward is flocks maintaining both exceptional productivity and robust metabolic health.
References are available on request.


